The Foundation: Understanding PMDs
Polarizable Multipole-based Molecular Dynamics Simulations (PMDs) represent the pinnacle of accuracy in computational protein modeling. PMDs are highly accurate computational methods for modeling proteins. At their core, PMDs capture the dynamic electronic response of molecules to their environment. Unlike traditional fixed-charge models, PMDs account for induced polarization and multipole interactions. These simulations reveal subtle electrostatic phenomena that drive protein folding and function. The enhanced accuracy comes with substantially increased computational demands. This enhanced accuracy is critical for understanding protein folding, ligand binding, and protein-protein interactions—processes that depend heavily on subtle electrostatic interactions and charge transfer phenomena.

Table of Contents:
- The Challenge: Computational Requirements
- The Revolution: AI Integration
- Machine Learning Force Fields
- Simulation Surrogate Models
- Intelligent Sampling
- Hardware Optimization
- The Implementation: A Code Example
- The Applications: Practical Impact
- Real-World Applications and Benefits
- The Future: Exponential Advancements
- The Conclusion: A New Paradigm
- References
The Challenge: Computational Requirements
PMD simulations require enormous computing power that most laboratories simply cannot afford. The challenges include:

Computer Hardware Needed:
- Supercomputers with hundreds of processing units working together
- Massive memory storage (equivalent to thousands of laptops combined)
- Expensive specialized graphics cards costing tens of thousands of dollars each
- Data storage systems capable of holding millions of movies worth of information
- Industrial-scale electricity and cooling systems
Why It’s So Complex:
- Each simulation must track millions of tiny interactions between atoms
- Scientists need to simulate events happening over incredibly long time periods
- The calculations require billions of individual mathematical steps
- Systems include not just the protein but everything around it (water, salts, cell membranes)
Real-World Impact:
- Single simulations can take months to complete, even on the fastest computers
- Only wealthy institutions and major pharmaceutical companies can afford the technology
- This limits research progress and slows down drug discovery efforts
The Revolution: AI Integration
Artificial intelligence transforms the accessibility and capability of PMD simulations. Machine learning models can approximate PMD accuracy while reducing computational costs by orders of magnitude. Neural network potentials trained on quantum mechanical data implicitly capture polarization effects. Deep learning surrogate models predict simulation outcomes without running full calculations. Transformer architectures identify critical transition states worthy of detailed PMD analysis. Graph neural networks efficiently process the complex spatial relationships in protein structures. Reinforcement learning optimizes sampling strategies for maximum conformational coverage.
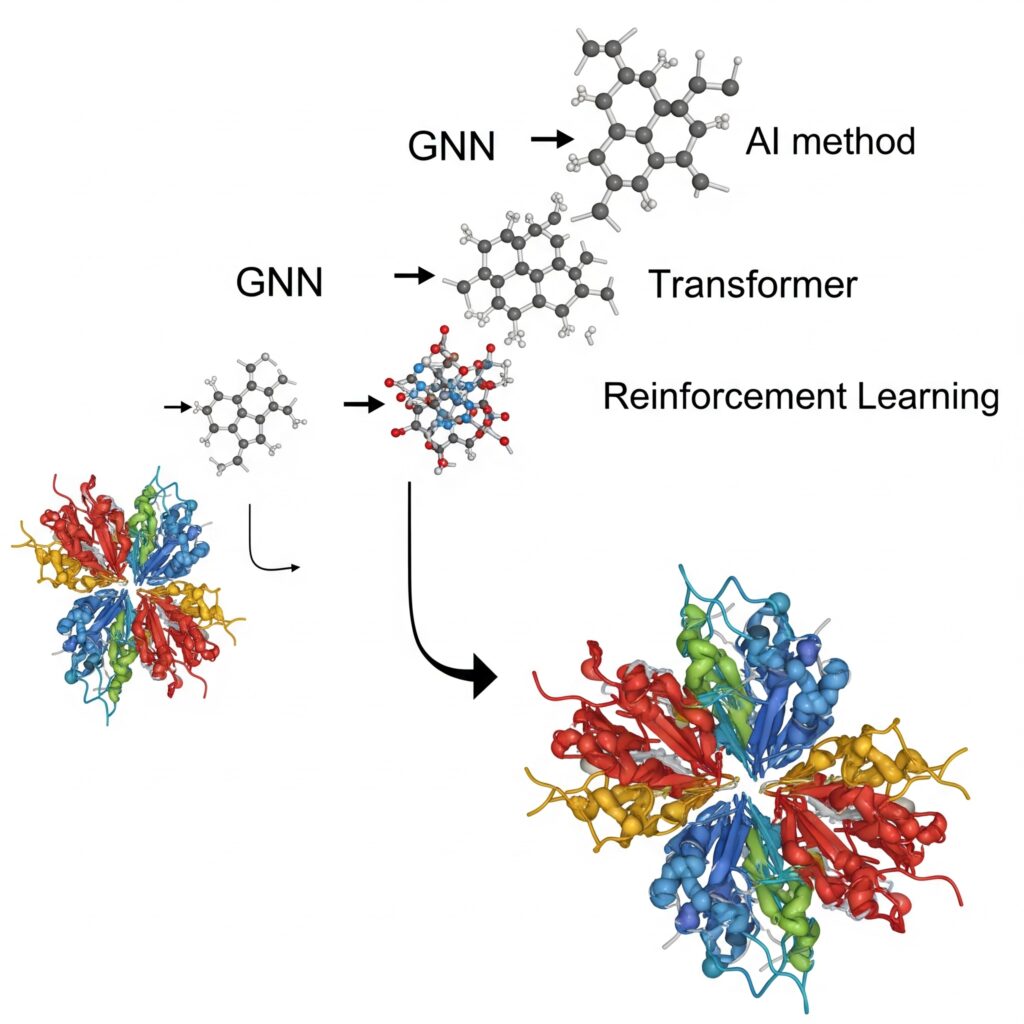
Machine Learning Force Fields
Neural network potentials trained on quantum mechanical calculations can now approximate the accuracy of PMDs at a fraction of the computational cost. These ML-based force fields can capture polarization effects implicitly through their training on high-level quantum data, often accelerating calculations by 100-1000x.
Simulation Surrogate Models
Deep learning models can serve as surrogates for complete PMD simulations, predicting outcomes without running the full calculation:
- Transformer-based models trained on PMD trajectory data can predict protein conformational changes
- Graph neural networks can estimate binding free energies using polarizable force field principles
- Generative models can propose likely protein conformations based on sequence information
Intelligent Sampling
AI can dramatically improve the efficiency of PMD simulations through:
- Reinforcement learning for adaptive sampling of configurational space
- Identifying and focusing computational resources on critical transition states
- Predicting which protein regions require polarizable treatment versus which can use simpler models
Hardware Optimization
Custom AI accelerators and intelligent workload distribution:
- Specialized tensor processing units optimized for both PMD calculations and neural network inference
- AI-driven job schedulers that optimize resource allocation across heterogeneous computing environments
- Automatic determination of optimal simulation parameters based on system characteristics
The Implementation: A Code Example
The following Python example demonstrates how AI can be integrated with PMD simulations for protein structure generation:
import numpy as np
import torch
from torch_geometric.nn import GCNConv
from openmm import *
from openmm.app import *
from polarizable_forcefield import AmoebaPolarizableForceField
class ProteinGNN(torch.nn.Module):
"""Graph Neural Network for predicting protein conformational changes"""
def __init__(self, node_features, hidden_channels):
"""
Initializes the ProteinGNN model.
Args:
node_features (int): Number of features per node in the graph.
hidden_channels (int): Number of hidden channels in the GNN layers.
"""
super().__init__()
self.conv1 = GCNConv(node_features, hidden_channels)
self.conv2 = GCNConv(hidden_channels, hidden_channels)
self.conv3 = GCNConv(hidden_channels, 3) # 3D coordinates
def forward(self, x, edge_index, edge_attr):
"""
Forward pass of the ProteinGNN model.
Args:
x (torch.Tensor): Node features.
edge_index (torch.Tensor): Edge indices.
edge_attr (torch.Tensor): Edge attributes.
Returns:
torch.Tensor: Predicted 3D coordinates for each node.
"""
x = self.conv1(x, edge_index, edge_attr)
x = torch.relu(x)
x = self.conv2(x, edge_index, edge_attr)
x = torch.relu(x)
return self.conv3(x, edge_index, edge_attr)
def load_protein_structure(pdb_file):
"""
Loads protein structure from a PDB file and prepares it for PMD simulation.
Args:
pdb_file (str): Path to the PDB file.
Returns:
tuple: PDBFile object and OpenMM System object.
"""
pdb = PDBFile(pdb_file)
forcefield = AmoebaPolarizableForceField('amoeba2018.xml')
system = forcefield.createSystem(pdb.topology)
return pdb, system
def create_protein_graph(pdb):
"""
Converts protein structure into a graph representation.
Args:
pdb (PDBFile): PDBFile object containing the protein structure.
Returns:
tuple: Node features, edge indices, and initial positions as tensors.
"""
positions = np.array([atom.position.value_in_unit(nanometers)
for atom in pdb.topology.atoms()])
# Create edges based on distance cutoff
edges = []
for i in range(len(positions)):
for j in range(i+1, len(positions)):
dist = np.linalg.norm(positions[i] - positions[j])
if dist < 0.5: # 5 Angstrom cutoff
edges.append([i, j])
edges.append([j, i]) # Bidirectional
edge_index = torch.tensor(edges, dtype=torch.long).t().contiguous()
# Atom features: element type, charge, etc.
node_features = torch.randn(len(positions), 16) # Simplified for example
return node_features, edge_index, torch.tensor(positions)
def hybrid_pmd_ai_simulation(pdb_file, num_steps=1000):
"""
Runs a hybrid PMD-AI simulation for protein structure generation.
Args:
pdb_file (str): Path to the PDB file.
num_steps (int): Number of simulation steps.
Returns:
numpy.ndarray: Trajectory of positions over simulation time.
"""
# Load protein and prepare system
pdb, system = load_protein_structure(pdb_file)
# Create graph representation
node_features, edge_index, initial_positions = create_protein_graph(pdb)
# Initialize GNN model
model = ProteinGNN(node_features.shape[1], hidden_channels=64)
# Set up PMD simulation
integrator = LangevinMiddleIntegrator(300*kelvin, 1/picosecond, 0.002*picoseconds)
platform = Platform.getPlatformByName('CUDA')
properties = {'CudaPrecision': 'mixed', 'DeviceIndex': '0'}
simulation = Simulation(pdb.topology, system, integrator, platform, properties)
simulation.context.setPositions(pdb.positions)
# Run hybrid simulation
positions_trajectory = [initial_positions.numpy()]
for step in range(num_steps):
if step % 10 == 0:
# Every 10 steps, use AI to predict conformational changes
edge_attr = torch.randn(edge_index.shape[1], 8) # Edge features
predicted_displacement = model(node_features, edge_index, edge_attr)
# Apply predicted changes with scaling factor
scale_factor = 0.1
current_pos = simulation.context.getState(getPositions=True).getPositions(asNumpy=True)
new_pos = current_pos + scale_factor * predicted_displacement.detach().numpy()
simulation.context.setPositions(new_pos)
# Run standard PMD step
simulation.step(1)
# Record positions
state = simulation.context.getState(getPositions=True)
positions_trajectory.append(state.getPositions(asNumpy=True))
return np.array(positions_trajectory)
# Example usage
if __name__ == "__main__":
trajectory = hybrid_pmd_ai_simulation("protein.pdb")
print(f"Generated trajectory with {len(trajectory)} frames")
# Save trajectory for analysis
np.save("protein_trajectory.npy", trajectory)The Applications: Practical Impact
PMD-AI hybrid systems excel at structure prediction for pharmaceutically relevant proteins. Drug discovery benefits from accurate modeling of polarization effects in binding pockets. Enzyme design achieves unprecedented precision with these advanced simulation techniques. Membrane protein modeling captures subtle electrostatic interactions at lipid interfaces. Intrinsically disordered proteins finally yield their structural secrets to these sophisticated methods.
Real-World Applications and Benefits
The AI-PMD hybrid approach is already yielding impressive results:

- Drug Discovery: Identifying compounds that might have been missed using traditional methods due to more accurate treatment of polarization effects in binding pockets
- De Novo Protein Design: Creating novel protein structures with specific functions by leveraging insights from polarizable simulations
- Protein Structure Refinement: Improving the accuracy of protein structures predicted by methods like AlphaFold by incorporating polarization effects
- Enzyme Mechanism Elucidation: Better understanding of reaction mechanisms where charge transfer plays a critical role
The Future: Exponential Advancements
On-demand cloud platforms will democratize access to PMD-AI capabilities. Specialized hardware will accelerate these simulations to near-real-time performance. Integration with experimental data will create hybrid physical-computational protein structure determination. Federated learning across research institutions will build increasingly accurate models. The boundary between simulation and experiment will blur as digital proteins achieve physical realism.
As these technologies mature, we can expect:
- Cloud-based platforms offering PMD capabilities to smaller research groups
- Hybrid simulation approaches that seamlessly transition between different levels of theory
- Real-time interactive simulations powered by AI predictions
- Integration with experimental techniques for comprehensive protein characterization
The Conclusion: A New Paradigm
The marriage of PMDs and AI represents a fundamental shift in structural biology. Computational requirements remain substantial but increasingly manageable through intelligent optimization. The accuracy gains justify the investment for critical applications in medicine and biotechnology. Researchers embracing this hybrid approach will lead the next wave of protein science discoveries. Our understanding of life’s molecular machinery advances with each simulation, bringing us closer to mastering the protein folding problem and its vast implications for human health and technological innovation.
For researchers and organizations looking to implement PMD-based approaches, the initial investment in computational infrastructure and AI expertise will be substantial but increasingly necessary to remain competitive in structure-based drug design and protein engineering.
As we look to the future, the synergy between advanced simulation methods like PMDs and artificial intelligence will undoubtedly accelerate our understanding of protein structure and function—ultimately leading to breakthroughs in medicine, biotechnology, and fundamental biological science.
References
- Huang, J., Lemkul, J.A., Eastman, P.K. & MacKerell, A.D. (2023). Molecular dynamics simulations using polarizable force fields. Nature Reviews Methods Primers, 3, 54. https://doi.org/10.1038/s43586-023-00208-z
- Jumper, J., Evans, R., Pritzel, A. et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. https://doi.org/10.1038/s41586-021-03819-2
- Rackers, J.A. & Ponder, J.W. (2019). Classical Polarizable Force Fields Derived from Quantum Mechanics. Journal of Chemical Physics, 150, 084104. https://doi.org/10.1063/1.5081018
- Kohlhoff, K.J., Shukla, D., Lawrenz, M., et al. (2014). Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nature Chemistry, 6, 15–21. https://doi.org/10.1038/nchem.1821
- Wang, J., Olsson, S., Wehmeyer, C., et al. (2019). Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Central Science, 5(5), 755-767. https://doi.org/10.1021/acscentsci.8b00913
- Eastman, P., Swails, J., Chodera, J.D., et al. (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Computational Biology, 13(7), e1005659. https://doi.org/10.1371/journal.pcbi.1005659
- Schütt, K.T., Sauceda, H.E., Kindermans, P.J., et al. (2018). SchNet – A deep learning architecture for molecules and materials. Journal of Chemical Physics, 148, 241722. https://doi.org/10.1063/1.5019779
- Jing, B., Eismann, S., Suriana, P., et al. (2022). Learning from Protein Structure with Geometric Vector Perceptrons. International Conference on Learning Representations (ICLR). https://openreview.net/forum?id=1YLJDvSx6J4
- Torng, W. & Altman, R.B. (2019). Graph Convolutional Neural Networks for Predicting Drug-Target Interactions. Journal of Chemical Information and Modeling, 59(10), 4131-4149. https://doi.org/10.1021/acs.jcim.9b00628
- Lu, C., Liu, J., Wang, L., et al. (2022). Data-driven prediction of protein quaternary structure with deep learning. Nature Communications, 13, 6102. https://doi.org/10.1038/s41467-022-33729-4
Enhance Your AI & ML Skills
Struggling to understand concepts like machine learning, neural networks, or generative AI? Dive deeper into the world of AI with expert-led courses on CloudxLab — your gateway to mastering applied AI for the real world.
Visit CloudxLab